The Power Drain of Aging: Mitochondria Under Stress
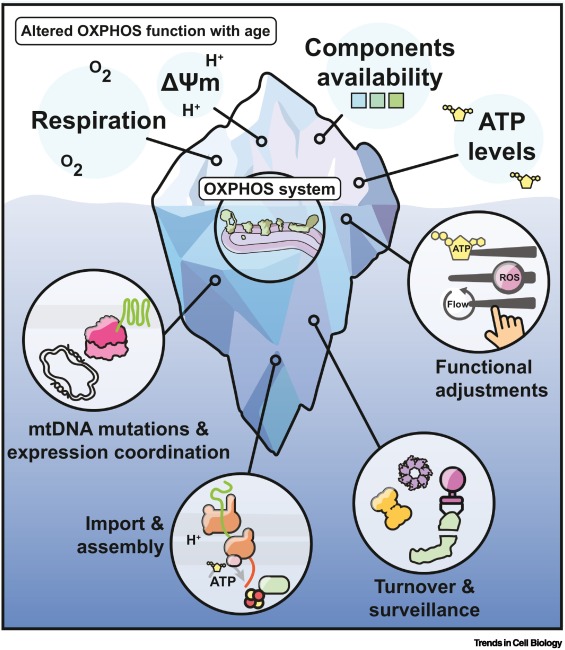
A new comprehensive paper published in Trends in Cell Biology by Hanna Salmonowicz et al, reveals how mitochondrial respiratory complexes—integral to OXPHOS—degrade with age. From genetic miscommunication to protein misfolding, from toxic ROS buildup to reduced energy production, our mitochondria face a full-blown identity crisis as we grow older. This dysfunction not only accelerates aging itself but also fans the flames of diseases like Alzheimer’s, heart failure, and sarcopenia.
Key Points of the Paper:
- Mitochondrial dysfunction is a hallmark of aging, primarily due to defects in the oxidative phosphorylation (OXPHOS) system.
- OXPHOS comprises five multi-subunit respiratory complexes (CI–CV) that are essential for ATP production, ROS generation, and metabolic signaling.
- Aging impairs multiple stages of the OXPHOS lifecycle, including:
- Coordination of nuclear and mitochondrial gene expression
- Import and processing of nuclear-encoded proteins
- Assembly of functional complexes
- Turnover and degradation of damaged complexes
- mtDNA mutations accumulate with age, primarily due to replication errors, leading to reduced OXPHOS efficiency and tissue-specific pathologies.
- Proteostasis and quality control mechanisms (e.g., mitochondrial proteases, mitophagy) are essential to maintain OXPHOS integrity and become challenged during aging.
- Aging can alter OXPHOS complex composition and organization, including the formation of supercomplexes and isoform switching
- Reactive oxygen species (ROS) have dual roles—both damaging and signaling—and are tightly linked to OXPHOS activity and dysfunction.
- Tissue and cell-type heterogeneity defines how OXPHOS alterations manifest during aging; for example, postmitotic, energy-demanding tissues like heart and brain are especially vulnerable.
- Lifestyle factors (e.g., physical inactivity, inflammation, hypoxia) directly impact OXPHOS dynamics in aging.
- Outstanding questions remain about the threshold of stress that triggers OXPHOS remodeling, the balance between beneficial and harmful adaptations, and how findings in models translate to humans.
Conclusion
- OXPHOS decline is central to age-associated mitochondrial dysfunction, yet the process is multi-layered, dynamic, and context-dependent.
- The accumulation of mtDNA mutations, disruption in gene expression coordination, and proteostatic decline are key vulnerabilities.
- New experimental tools (e.g., high-throughput multi-omics, mitochondrial genome editing, cryo-EM) are critical for uncovering the fine regulation of OXPHOS in aging.
Despite decades of research, whether OXPHOS dysfunction is a cause or consequence of aging remains unresolved.
A comprehensive, tissue-specific understanding of OXPHOS changes will pave the way for interventions targeting mitochondrial health in age-related diseases.